




Super-resolution microscopy is on the verge of revolutionizing our understanding of cell biology. Our motivation is to develop imaging tools that allow us to visualize the full complexity of a cell and its molecular architecture. With this technology, we aim to understand how protein assemblies form, function, and degrade in the environment of a cell.
Optical super-resolution microscopy for structural cell biology
We aim to render super-resolution fluorescence microscopy into an “optical omics” technology. To achieve this aim, we develop methods for biomolecule labeling and advanced theoretical models and image analysis tools to enable super-resolution microscopy in cell biology samples.
We currently address the following topics:
- Multiplexing. Biomolecules have many interaction partners in a cell and are embedded in a complex environment. We aim to decipher the structural organization of biomolecular “networks” in cells by developing multi-target labeling for single-molecule localization microscopy (SMLM), stimulated emission depletion (STED) microscopy, and other imaging methods.
- Molecular quantification. The function of biomolecules is encoded in the composition and structural organization of protein assemblies. We use single-molecule analysis methods to quantify protein stoichiometries of such assemblies in their native environment, the cellular environment.
- High-speed imaging. We explore software-assisted approaches to minimize the imaging time while conserving the image quality and information content. We apply these tools for for live cell imaging and multiplexed super-resolution imaging in fixed samples.
- Minimize photobleaching. Photobleaching is a major obstacle in (super-resolution) fluorescence microscopy. We develop labeling tools and computational methods that minimize photobleaching while conserving the information content of fluorescence images.
Protein assemblies in membranes
Membranes are essential for the functionality of cells as entities and within a multi-cellular environment. The plasma membrane constitutes a boundary separating the cell interior from its environment. Several intracellular organelles are compartmentalized through membranes. Within and at membranes, proteins together with lipids assemble into essential cellular reaction hubs. Membrane receptors communicate information across these boundaries. We are interested in various types of membrane proteins and aim to understand the functional principles that underlie the formation, function and degradation of membrane-associated protein assemblies in a cell.
Membrane receptors. Membrane receptors constitute the communication interface of a cell to its environment, and translate extracellular signals into cellular responses. Using optical imaging in live and fixed cells, we aim to understand how the spatial organization and dynamics determine the function of membrane receptors and associated protein assemblies.
Many-target labeling for (super-resolution) fluorescence microscopy
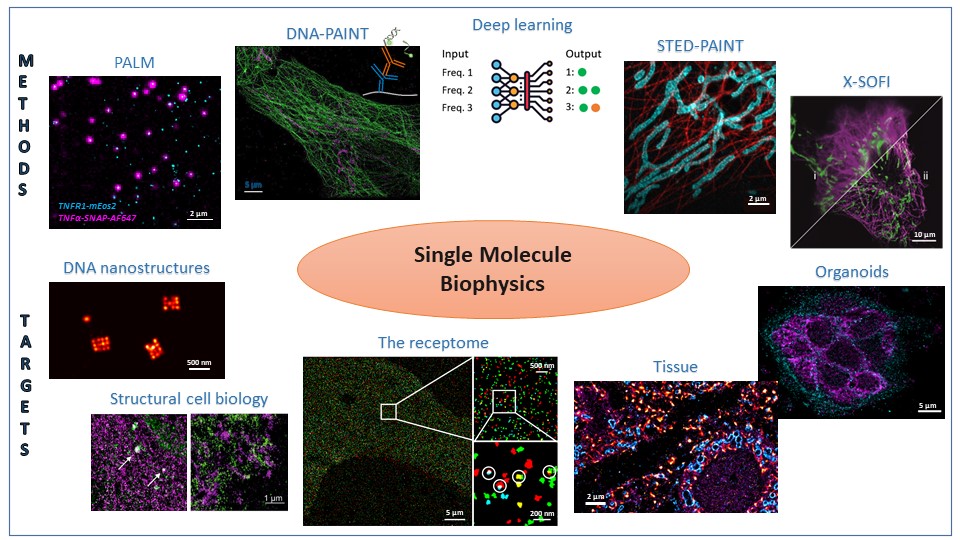
DNA-labeled probes
In DNA-assisted SMLM (DNA-PAINT), a single-molecule fluorescence signal is generated from the transient hybridization of a fluorophore-labeled oligonucleotide (imager strand) to a sequence-complementary docking strand attached to a target molecule. In consequence, the “color” is encoded in the oligonucleotide sequence. This increases the number of addressable targets compared to multi-target imaging with spectrally distinct fluorophores. We employ DNA-PAINT for SMLM imaging of multi-protein networks of membrane proteins, of subcellular organelles such as the ER and the Golgi apparatus, and in neuronal brain tissue.
|
|

| Figure 1: Multi-target/Multiplexed SMLM using DNA-labeled antibodies (for more information, please see (Schröder et al. 2020)). |
We transfer the concept of DNA-labeled protein labels to other (super-resolution) fluorescence methods, such as STED, super-resolution optical fluctuation imaging (SOFI), and confocal microscopy.
Molecular quantification of protein complex stoichiometries from super-resolution imaging data
Quantitative PALM/dSTORM
Proteins organize into assemblies with specific stoichiometries that determine their function. This often dense packaging of proteins cannot be resolved in situ with to-date optical super-resolution imaging methods. To bypass this limitation, we developed kinetics-based image analysis of SMLM data recorded with photoswitchable fluorescent proteins/organic dyes. The kinetics of fluorescence “blinking” of fluorophores is directly related to molecular number. We apply quantitative PALM/dSTORM to determine the stoichiometry of membrane proteins in protein assemblies.
|
|
 |
| Figure 2: The photo-”blinking” kinetics of fluorescent proteins (A, B) are related to molecule numbers and can be modeled by kinetic equations (C). This in turn allows us to determine the stoichiometry of protein assemblies (for more information, please see (Dietz and Heilemann 2019)). |
This technique profits from stoichiometric labeling of target proteins in cells. For this purpose, we used CRISPR/Cas12a for endogenous labeling of membrane proteins with photoswitchable fluorescent proteins (Baldering et al. 2021).
|
|

|
Figure 3: Endogenous tagging of the MET receptor with mEos4b in HEK293T cells using the CRISPR/Cas12a technology (for more information, please see (Baldering et al. 2021)). |
Quantitative DNA-PAINT
In DNA-assisted SMLM (DNA-PAINT), a single-molecule fluorescence signal is generated from the transient hybridization of a fluorophore-labeled imager strand (diffusing in the imaging buffer) to a sequence-complementary, target-labeled docking strand. The high excess concentration of the imager strand in the imaging buffer provides a constant signal over long acquisition times. The kinetics of oligonucleotide hybridization and dissociation report on molecule numbers.
 |
|
Figure 4: Quantitative DNA-PAINT extracts molecule numbers from kinetic parameters of DNA hybridization (for more information, please see (Dietz and Heilemann 2019)). |
Increasing the imaging speed in live cell and super-resolution microscopy
Deep-learning assisted live cell imaging: fast, long acquisition times, minimal phototoxicity
The dynamics of subcellular structures occur on a large range of time scales. What is needed are tools for live-cell imaging that on the one hand provide a fast temporal resolution, and on the other hand allow for long-time observation of cellular dynamics. For this purpose, we employ neural networks that are trained to predict high-contrast fluorescence images from low-contrast data that were recorded by fast imaging or with low light exposure.
 |
|
Figure 5: A neural network predicts a high contrast fluorescence image from a low contrast image recorded with low illumination intensities. |
Deep-learning assisted SMLM
SMLM builds on the spatially separated detection of the fluorescence signal of single emitters, which makes it a rather slow method. We follow different strategies to increase the imaging speed in SMLM, and for example (1) integrate neural networks that reconstruct super-resolved images from high-density single-molecule data and (2) construct neural networks that discriminate overlapping emission signal of different types of fluorophores.
Minimizing photobleaching in (super-resolution) fluorescence microscopy
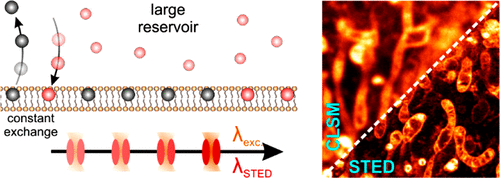
Replenish the label & keep the signal: exchangeable labels for fluorescence microscopy
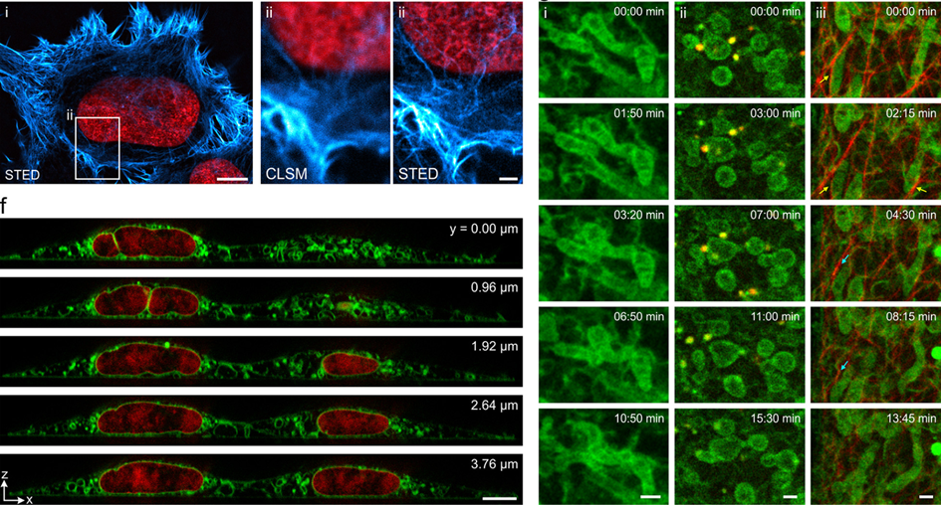
Fluorophore labels that transiently and repetitively bind to a target lead to a constant fluorescence signal over time. This in turn prevents a loss of signal because of photobleaching. Minimizing photobleaching with exchangeable labels in the first place extends the overall observation time. This allows optimizing signal-to-background/-noise, and facilitates multi-color, large-volume 3D and live cell imaging.
We apply this concept for super-resolution fluorescence methods such as STED, SMLM and SOFI, as well as for multi-color confocal microscopy.
 |
| |
 |
|
Figure 6: Exchangeable fluorophore labels minimize photobleaching in STED microscopy (top) and allow for multi-color, 3D and live cell imaging (bottom) (for more information, please see (Spahn et al. 2019)). |
Neural networks enable live-cell imaging with low intensities
Deep neural networks that were trained on noisy imaging data can generate high-quality fluorescence images from samples that were recorded using low light intensities. This enables long observation times in live cells, and at the same time limits phototoxicity.